Finding New Overlapping Genes and Their Theory (FOG Theory)
Das übergeordnete Ziel des Projekts „Finding new overlapping genes and their theory (FOG Theory)” ist es, neue überlappende proteinkodierende DNA-Sequenzen in Prokaryoten zu finden und zu verifizieren, die zugrunde liegenden Kodierungscharakteristika zu verstehen und ihren Ursprung und ihre Evolution mit Hilfe von Modellen aus der Informations- und Kommunikationstheorie zu untersuchen.
Im ersten Teil des Verbundprojekts konnte gezeigt werden, dass bakterielle Genome viele nicht-zufällige lange offene Leserahmen enthalten, bei denen es sich um überlappende Gene handeln könnte. Tatsächlich wurde eine Vielzahl von Kandidaten für überlappende Gene mit Hilfe von Datenanalysetechniken in mehr als 50 bakteriellen Genomen identifiziert. Experimentelle Arbeiten am Modellorganismus EHEC haben mehrere Transkriptionseinheiten aufgedeckt, bei denen es sich um überlappende Gene handeln könnte.
In der nächsten Periode wollen wir unser bisher erworbenes Wissen nutzen, um
- die Charakteristika und Besonderheiten überlappender Gene für die computergestützte Vorhersage und Expertenbewertung mittels Visualisierung zu identifizieren,
- die biologische Funktion ausgewählter Zielkandidaten überlappender Gene experimentell zu charakterisieren und
- Randbedingungen für den evolutionären Ursprung überlappender Gene zu etablieren.
Das Projekt „Finding new overlapping genes and their theory (FOG Theory)” ist ein Verbundprojekt dreier Gruppen: der Data Analysis and Visualization Group an der Universität Konstanz, des Instituts für Nachrichtentechnik und Angewandte Informationstheorie – TAIT der Universität Ulm und des Lehrstuhls für Mikrobiologie am Zentralinstitut für Ernährungs- und Lebensmittelforschung (ZIEL) der Technischen Universität München. Es ist Teil des Schwerpunktprogramms „Information and Communication Theory in Molecular Biology” (InKoMBio) der Deutschen Forschungsgemeinschaft (DFG).
Teilprojekt: Visual Analysis of Next-Generation-Sequencing Data
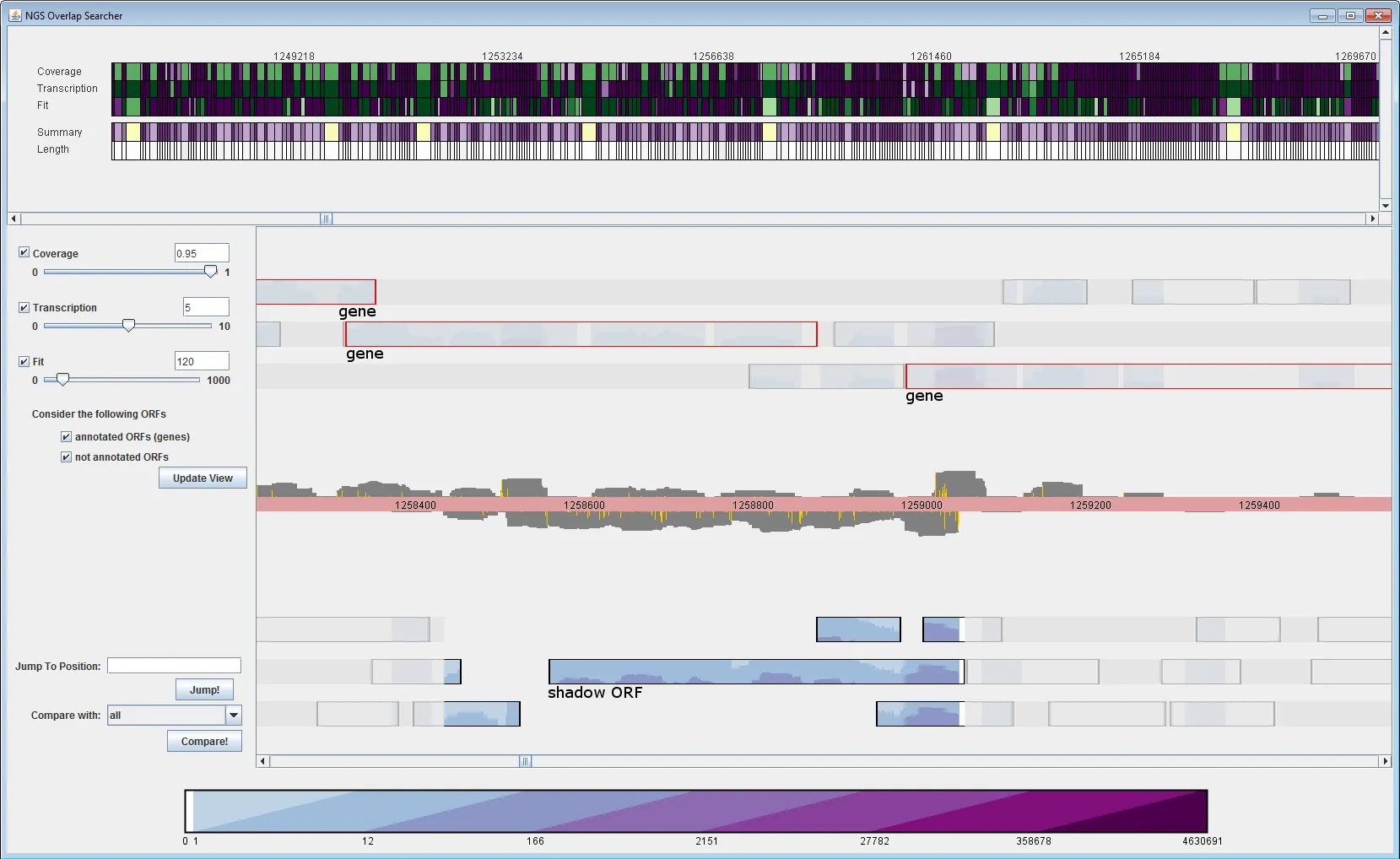
Next-Generation-Sequencing-Technologien (NGS) erlauben es, große Mengen an DNA-Sequenzen in kurzer Zeit und zu geringen Kosten zu sequenzieren. Die Technik wird nicht nur zur Sequenzierung ganzer Genome eingesetzt, sondern auch zur (indirekten) Sequenzierung der mRNA (des Transkriptoms) einer Zelle. mRNA wird von Genen transkribiert und kann als Bauplan eines Gens betrachtet werden, der zum Aufbau des von diesem Gen kodierten Proteins dient. Somit kann bei jeder transkribierten Genomregion vermutet werden, dass sie ein proteinkodierendes Gen enthält. Wird mRNA (indirekt) sequenziert, können diese Sequenzen zurück auf das Genom gemappt werden. Dies erlaubt nicht nur die Identifikation von Genen, die unter einer bestimmten Bedingung aktiv sind, sondern auch die Identifikation neuer Gene und sogar die Abschätzung der Menge transkribierter mRNA. Daher ist NGS die Methode der Wahl zur Identifikation neuer überlappender Gene.
Da NGS Längenbeschränkungen unterliegt, ist es nicht möglich, eine mRNA über ihre gesamte Länge zu sequenzieren. Die mRNA muss daher fragmentiert werden, um sequenziert zu werden. Da beim Sequenzieren dieser Fragmente jedoch Sampling-Effekte auftreten, führt das Mapping der sequenzierten Fragmente (sogenannter Reads) zu einer ungleichmäßigen Coverage über die transkribierte Region.
Neben den großen Datenmengen, die bei NGS anfallen (dies können mehr als eine Million Reads pro Experiment sein), stellen diese ungleichmäßigen Coverages somit eine ernsthafte Herausforderung bei der Analyse von NGS-Daten dar.
Es sind daher neuartige Methoden der Datenanalyse und Visualisierung erforderlich, um dem biologischen Experimentator das Verständnis der Ergebnisse zu ermöglichen. Insbesondere bei Kandidaten für überlappende Gene ist eine anschließende Begutachtung der Ergebnisse durch einen Experten notwendig.
Um die Analyse von Next-Generation-Sequencing-Daten, insbesondere im Hinblick auf Kandidaten für überlappende Gene, zu erleichtern, haben wir ein neues Visual-Analytics-System (VA) entwickelt. Dieses VA-System erlaubt es, interessante Regionen anhand einer benutzerdefinierten Interestingness-Funktion zu bestimmen. Darüber hinaus ermöglicht eine Genom-Übersichtsleiste das einfache Auffinden der interessantesten Fälle und unterstützt den Analysten beim Umgang mit der großen Datenmenge. Außerdem visualisieren wir die Transkriptions-Coverage in den ORF-Darstellungen (Open Reading Frame), um ein einfacheres Mapping der Transkriptions-Coverage auf einen ORF zu ermöglichen.
Diese Arbeit wurde teilweise von der Deutschen Forschungsgemeinschaft (DFG) im Rahmen des SPP 1395 (Information and Communication Theory in Molecular Biology, InKoMBio), Projekt „Finding new overlapping genes and their theory (FOG-Theory)”, gefördert.